Neuroblastoma
-
Essential Information
Summarize
Neuroblastoma (NB) does not have a high incidence, but is the most common tumor in infancy. It often occurs in children under 5 years of age, especially in infants under 2 years of age. Neuroblastoma is an embryonal malignant tumor of the sympathetic nervous system caused by adult nerve cells (immature nerve cells). In the developing embryo, these cells invade, migrate along the neuraxis, and populate the sympathetic ganglia, adrenal medulla, and other sites. The distribution pattern of these cells correlates with the site of primary neuroblastoma.
Neuroblastoma usually originates in the adrenal glands in the neural tissue, but can also originate in the sympathetic tissue near the abdomen, chest, spinal cord, or cervical spine.
The adrenal glands are located in the back of the upper abdomen, at the top of each kidney. The adrenal glands produce important hormones that help control heart rate, blood pressure, blood sugar, and the way the body responds to stress.
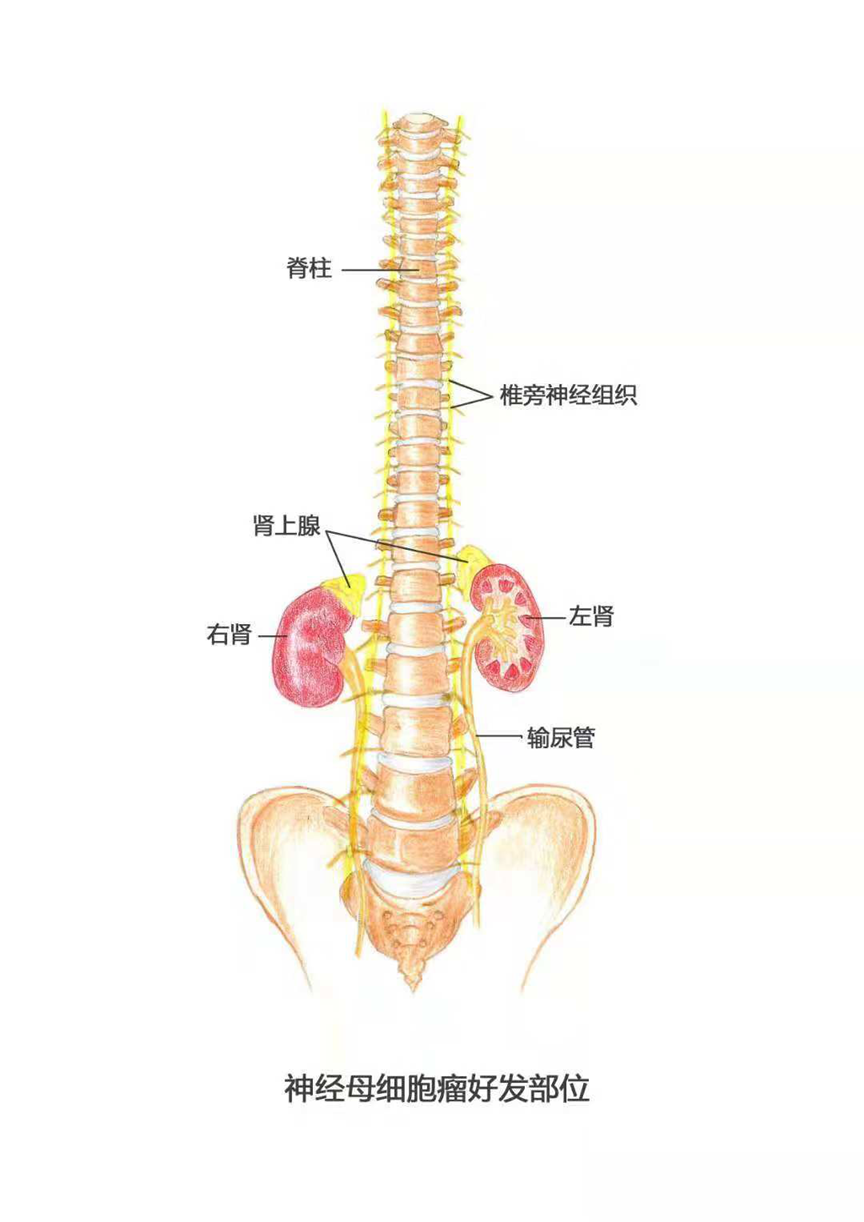
Figure 1 Neuroblastoma is seen in the adrenal and paravertebral nerve tissue from the neck to the pelvis
Epidemiological
incidence of a disease
Neuroblastoma accounts for 6% of all childhood cancers in the United States. Approximately 800 new cases are diagnosed each year in the United States. According to the Surveillance, Epidemiology, and End-Report (SEER), the incidence rate is approximately 10.54 cases per million children. Incidence rates in other industrialized countries appear to be similar to those observed in the United States. International reports indicate that the incidence of neuroblastoma is highest in high-income countries in Europe and North America, and lower in low-income countries in Africa, Asia, and Latin America, with more detailed data on Asia unavailable.
80% of patients are younger than 5 years of age at the time of diagnosis, and the median age at diagnosis is 19 months. The disease is rarer in people over 10 years of age.
Etiolog & Risk factors
risk factor
Neuroblastoma begins with adult nerve cells, which are immature nerve cells created during fetal development. As the fetus matures, the adult nerve cells eventually change into nerve cells, fibers, and cells that make up the adrenal glands. Most adult nerve cells mature and develop into nerve cells when the fetus is born. However, a small number of immature nerve-forming cells are present in some newborns. In most cases, these nerve-forming cells develop into nerve cells as the infant's nervous system matures. In affected infants, however, the mutated nerve cells do not mature properly and instead grow and divide, eventually leading to neuroblastoma.
The cause of the transformation of adult nerve cells into neuroblastoma is unknown, and no specific environmental exposures or risk factors have been identified.
1. Family history
Patients with certain familial genetic disorders, such as Hirschsprung's disease or central congenital hypoventilation syndrome, carry mutations in the Phox2B gene associated with the development of neuroblastoma.
Also, certain family histories of neuroblastoma are associated with inherited mutations in the ALK gene. In addition, familial deletions of chromosome 1p36 or 11q14-23 loci are also responsible for familial neuroblastoma.
Children with a family history of neuroblastoma may be more susceptible to the disease and usually develop it earlier (average age of onset is 9 months). However, only 1-2% of neuroblastoma patients have a family history, and the vast majority of neuroblastoma cases are not due to genetic causes.
- genetic mutation
Studies have shown that people with specific variants in genes such as TP53, NRAS, BRCA2, PHOX2B, and ALK, as well as those with deletions on chromosome 1p36 or at the 11q14-23 locus, have a greater chance of developing neuroblastoma. It is uncertain exactly what causes the initial gene mutation in neuroblastoma.
Classification & Stage
Currently, there are two systems used internationally for neuroblastoma staging: the International Neuroblastoma Risk Group (INRG) staging system and the International Neuroblastoma Staging System (INSS).The INRG staging is a pre-treatment staging system based on Imaging Risk Factors (IDRFs); while the INSS is a postoperative staging system based on surgical situation and pathology.
International Neuroblastoma Risk Group (INRG) staging system
INRG stages neuroblastoma primarily based on relevant risk factors defined by imaging (e.g., ultrasound, CT, MRI, etc.) in the following stages:
Phase L1
Tumor only at the primary site and no imaging-defined risk factors. (Imaging-defined risk factors are a series of risk factors that are visible by imaging, such as whether the tumor surrounds an artery and infiltrates adjacent soft tissues and organs.)
L2
Absence of distant metastases and presence of imaging-defined risk factors.
Phase M
Have distant metastases (except MS stage).
MS period
Patients were diagnosed at an age of less than 18 months, and the tumor had metastasized only to the liver, skin, or bone marrow (no more than 10% of bone marrow cells were invaded by the cancer, along with no metastases to either bone or bone marrow on the MIBG scan).
International Neuroblastoma Staging System (INSS)
The INSS, on the other hand, classifies neuroblastomas into the following stages based on the extent to which the tumor can be surgically resected and the postoperative pathology:
Phase 1
The tumor can be completely removed surgically (although a small number of residual cancer cells may still be found at the margins of the tissue after surgery under the microscope) and has not spread to nearby lymph nodes (but may have invaded the lymph nodes surrounded by the tumor).
Phase 2A
The tumor has developed only locally at the site of origin, but surgery cannot completely remove the tumor because of its size, location, or proximity to other organs. The tumor has not spread to nearby lymph nodes (but may have invaded lymph nodes surrounded by the tumor).
Phase 2B
Tumors that have developed only locally at the site of origin, but cannot always be completely removed by surgery. The tumor has metastasized to lymph nodes on the same side of the body, but not yet to lymph nodes on the opposite side of the body (i.e., the metastasis has not crossed the midline of the spine).
Phase 3
The tumor has not yet developed distant metastases but meets one of the following conditions: the tumor has crossed the midline of the spine and cannot be removed surgically and may have spread to nearby lymph nodes; or the tumor is still in the area of origin but there are metastases to the contralateral lymph nodes; or the tumor originated in the midline area of the body and has spread to both sides and cannot be removed surgically.
Phase 4
The tumor has metastasized to lymph nodes, bone marrow, bone, liver, or other organs in distant parts of the body (except stage 4S).
Phase 4S
The patient was diagnosed at an age of less than 1 year and the tumor developed only locally, (stage 1 and 2), but metastases occurred in specific sites (e.g., liver, skin, bone marrow, etc.) and there were no bone metastases. If bone marrow metastasis has occurred, no more than 10% of the bone marrow cells have been invaded by the cancer cells, while there are no metastases in either bone or bone marrow on MIBG scan.
Risk classification based on INRG staging
The INSS, on the other hand, classifies neuroblastomas into the following stages based on the extent to which the tumor can be surgically resected and the postoperative pathology:
very low risk group
● INRG stage L1 or L2 with histology showing an imminent mature ganglioneuroma or mixed ganglioneuronal neuroblastoma.
● INRG stage L1, histologic examination of a pathologic type other than ganglioneuroma and mixed ganglioneuronal neuroblastoma, and absence of MYCN gene amplification.
● INRG staging MS, children younger than 18 months of age at diagnosis, without MYCN gene amplification and without chromosome 11q aberrations.
low-risk group
● INRG stage L2, children younger than 18 months of age at diagnosis, histology of a pathology other than ganglioneuroma and mixed ganglioneuroblastoma, absence of MYCN gene amplification and absence of chromosome 11q aberrations.
● In INRG stage L2, the patient is older than 18 months of age at diagnosis, histologically shows differentiated neuroblastoma or nodular ganglioneuroblastoma (differentiated within the nodes), has no MYCN gene amplification, and has no chromosome 11q aberrations.
● INRG stage M. The child is younger than 18 months of age at diagnosis and has no MYCN gene amplification or chromosomal hyperdiploidy.
medium risk group
● INRG staging L2, the child was diagnosed at an age younger than 18 months, there was no MYCN gene amplification, and there was a chromosome 11q aberration.
● INRG stage L2, patients were diagnosed at an age greater than 18 months, histology showed differentiated neuroblastoma or nodular ganglion cell neuroblastoma (differentiated within the nodes) without MYCN gene amplification and with a chromosome 11q aberration.
● INRG staging L2, patient's age at diagnosis >18 months, histology showing undifferentiated or poorly differentiated neuroblastoma or nodular ganglioneuronal neuroblastoma (undifferentiated or poorly differentiated type within the nodes) without MYCN gene amplification.
● INRG stage M. The child was diagnosed at an age of less than 12 months, had no MYCN gene amplification, and had diploid chromosomal ploidy.
high risk group
● INRG stage L1 with histologic examination of a pathologic type other than ganglioneuroma and mixed ganglioneuronal neuroblastoma with MYCN gene amplification.
● INRG staging L2 with MYCN gene amplification.
● INRG stage M. The child was diagnosed at an age of less than 18 months and had an amplification of the MYCN gene.
● INRG stage M. Patients were diagnosed at an age greater than 18 months.
● INRG staging MS stage, children diagnosed at less than 18 months of age, without MYCN gene amplification but with chromosome 11q aberrations.
● INRG staging MS stage, children diagnosed at less than 18 months of age with MYCN gene amplification.
Hazard classification based on INSS staging
low-risk group
● All cases in INSS Stage 1.
● INSS stage 2A or 2B, no MYCN gene amplification, and surgical resection of 50% or more of the tumor.
● INSS stage 4S, the child was diagnosed at an age of less than 12 months, there was no MYCN gene amplification, histopathology was favorable, and the chromosomal ploidy was hyperdiploidy.
medium risk group
● INSS stage 2A or 2B, no MYCN gene amplification, and surgery can only remove less than 50% of the tumor.
● INSS stage 3, children diagnosed at less than 18 months of age, no MYCN gene amplification.
● INSS stage 3, children were diagnosed at an age greater than 18 months, there was no MYCN gene amplification, and the histopathology was favorable.
● INSS stage 4, children diagnosed at less than 12 months of age, no MYCN gene amplification.
● In INSS stage 4, the children were diagnosed between 12 and 18 months of age, had no MYCN gene amplification, had hyperdiploidy, and had favorable histopathology.
● INSS stage 4S, no MYCN gene amplification, unfavorable histopathology or diploid chromosomal ploidy.
high risk group
● INSS stage 2A or 2B with MYCN gene amplification.
● INSS stage 3 with MYCN gene amplification.
● INSS stage 3, children diagnosed at age greater than or equal to 18 months, no MYCN gene amplification, and unfavorable histopathology.
● INSS stage 4, children diagnosed at less than 12 months of age with MYCN gene amplification.
● In INSS stage 4, children were diagnosed between 12 and 18 months of age, had an amplification of the MYCN gene, or had diploid chromosomal ploidy, or had unfavorable histopathology.
● INSS stage 4, children diagnosed at age >18 months.
● INSS stage 4S with MYCN gene amplification.
Clinical manifestations
Signs and symptoms of neuroblastoma correlate with where the tumor originates, the size of the tumor, its growth, and whether it has spread. Common symptoms include bloating, abdominal pain, constipation or diarrhea, vomiting, weight loss, anorexia, dyspnea, skin lumps, fatigue, and bone pain, as well as rare symptoms such as hypertension, chronic diarrhea (secondary to vasoactive intestinal peptide secretion), and ophthalmoscopic tremor-myoclonus syndrome (OMS).
Because early symptoms of neuroblastoma are inconspicuous and varied, many children are already suffering from tumor metastasis by the time they seek medical attention. Therefore, for children (especially those under 5 years of age), parents need to pay more attention to the symptoms of unexplained abdominal pain, chest pain, subcutaneous lumps, and orbital bruises, and take their children to the doctor promptly.
flank
About 65% of tumors originate in the abdomen, and children may find an asymptomatic abdominal mass when their parents or caregivers give them care.
If a tumor in the abdomen grows expansively and affects the visceral nervous system, the child may experience abdominal pain or not feel hungry all the time, or even lose weight as a result.
The tumor may also affect the child's bowel patterns, and the child may experience constipation or diarrhea. Some children with tumors located in the pelvis may also have difficulty urinating.
If the tumor grows in the lower abdomen or pelvic area, it may also cause edema in the legs. In boys, it may cause scrotal edema.
chest and neck
A tumor in the neck is usually visible from the outside as a hard lump that is usually not painful when pressed. Neuroblastoma in the neck sometimes presses on the trachea or esophagus, causing your child to cough, have trouble breathing, or difficulty swallowing.
Tumors in the chest are not easy to detect on the outside and may cause shortness of breath, coughing, or difficulty breathing. Chest tumors that extend into the neck can compress the sympathetic nerves and cause Horner's syndrome.
Horner syndrome is also known as pediatric cervical sympathetic paralysis syndrome, Bernard-Horner syndrome, and Claude-Bernard-Horner syndrome. The cause of the disease is that the tumor compresses or destroys the nerve pathway from the sympathetic nerve center to the eye, resulting in symptoms such as ptosis, pupil shrinkage, eyeball inversion, and little or no sweat on the face on one side. Usually, the child's eyelid is drooping unilaterally and cannot be lifted, and the pupil (the dark black dot in the center of the black eye) of each eye appears to be large or small.
In some children, neuroblastoma in the chest may compress blood vessels, causing localized venous or lymphatic reflux obstruction, resulting in swelling of the face, neck, arms, and chest, sometimes causing red ecchymosis on the skin.
central nervous system, CNS
When the tumor grows next to the spine, especially into the spinal canal, it may affect the sensation and mobility of the limbs, causing the child to lose sensation in the arms or legs and feet, or to have reduced mobility in the arms or legs and feet.
If the tumor grows inside the skull, the child may experience headaches, dizziness, vomiting, blurred vision or even impaired consciousness.
"Blueberry Muffin Babies."
Within the first few months of neuroblastoma, blue or purple bumps that look like small blueberries are sometimes seen on your child's skin, a sign that the cancer may have spread to the skin. The condition is easy to treat and usually shrinks or disappears on its own.
Paraneoplastic Syndrome
In some patients with malignant tumors, although the tumor has not yet metastasized, the chemicals secreted by the tumor have affected organs outside the primary lesion, known as paraneoplastic syndrome. Very few children with neuroblastoma develop rapid eye movements and coordination difficulties (nystagmus-myoclonus syndrome), severe diarrhea, or high blood pressure.
lymphatic node
If neuroblastoma has metastasized, then the most common site of metastasis is the lymph nodes, which are often enlarged in children. Specifically, enlarged lymph nodes can often be felt in the child's neck, clavicle, armpit, groin and other parts of the body, which feel almost like small lumps under the skin.
Bone and bone marrow
If neuroblastoma metastasizes to the bones, it can cause severe pain, and the child may have trouble walking or may be reluctant to walk.
The tumor may also metastasize to the bone marrow. At this point, the child may experience fever, fatigue, weakness, susceptibility to infection, and sometimes difficulty in clotting, and a very small contusion or cut can cause large bruises or bleeding.
Clinical Department
not have
Examination & Diagnosis
diagnostic strategy
Neuroblastoma is usually seen in pediatrics, pediatric surgery or oncology. It is difficult to make an early diagnosis of this disease. When a patient begins to show symptoms, the doctor relies on blood and urine tests, imaging tests such as CT or magnetic resonance imaging (MRI) of the chest and abdomen, functional imaging tests, bone marrow tests, and biopsy specimens, in addition to an aggressive history and physical examination, to confirm the diagnosis of neuroblastoma. If the questioning reveals that the patient's age, signs and symptoms, and other characteristics support the possible presence of neuroblastoma, the doctor will perform further biochemistry, imaging, and biopsy.
In addition, because neuroblastoma exhibits atypical signs and symptoms, neck tumors need to be differentiated from lymphadenopathy, and mediastinal and retroperitoneal tumors need to be differentiated from germ cell tumors and kidney tumors.
Confirmation criteria (one of the following two)
1. Clear histopathological diagnosis of tumor tissue under light microscope.
2. Bone marrow aspiration biopsy or ring-drill biopsy showing evidence of bone marrow metastases, along with elevated urinary or serum catecholamine or their metabolite levels.
biochemical examination
This consists mainly of blood and urine tests for catecholamines.
The body produces many different types of hormones. Sympathetic nerve cells usually release hormones called catecholamines, which enter the bloodstream and are eventually broken down into smaller fragments called metabolites, the two main ones being vanillylmandelic acid (VMA) and homovanillic acid (HVA).VMA and HVA are usually excreted in the urine.
Neuroblastoma cells can also produce catecholamines. A diagnosis of neuroblastoma is supported if the levels of VMA and HVA in the urine or blood are higher than expected.
If neuroblastoma is suspected or has been found, routine blood, liver and kidney function, and electrolyte profile tests are usually performed at the same time.
Imaging
Imaging tests such as ultrasound, X-ray, CT, and MRI are helpful in the diagnosis of neuroblastoma, the extent of tumor spread, and the evaluation of the effectiveness of tumor treatment. The commonly used imaging tests are:
:: Ultrasound: Because ultrasound is quick, easy to perform and non-radioactive, it is generally used as one of the first tests for patients with suspected tumors. It is often used to look for tumors of abdominal origin.
● CT: A CT scan is often used to look for neuroblastoma in the abdomen, pelvis, and chest and can estimate the size of the tumor.
● Magnetic Resonance Imaging (MRI): MRI is better than CT for viewing the brain and spinal cord, is not radioactive, and can also estimate the size of tumors.
● MIBG Scan: Usually used to detect how far a neuroblastoma has spread. It is usually done after a CT or MRI has been completed.MIBG is similar to norepinephrine in that after it is injected into a vein through the bloodstream, in most patients it will attach to neuroblastoma cells anywhere in the body. After 1 to 3 days, scanning the body with a special camera will help the doctor know where the neuroblastoma is and whether it has spread to the bones, bone marrow, and/or other parts of the body.
● Bone scan: To help check whether neuroblastoma has spread to the bones, it is now mostly replaced by PET-CT or MIBG, and the need for this test can be determined by the results of the MIBG.
PET-CT scan: A combination of a PET scan and a CT scan. Although PET scan is not as detailed as CT and MRI, it can directly scan the whole body and is usually used as an auxiliary reference to CT.PET-CT scan is usually used in patients with negative MIBG results to check the spread of tumors.
● Chest radiograph (X-ray): Chest radiographs are routinely performed when patients present with symptoms of common chest primaries, such as dyspnea, but by themselves are of limited diagnostic utility for neuroblastoma and are not as clear as CT or MRI.
Biopsy and pathology
Although biochemical and imaging tests can effectively assist in the diagnosis of neuroblastoma, tissue biopsy and pathology are the indicators of a definitive diagnosis of the disease.
Tumor tissue specimens were obtained by open surgery for direct biopsy.
If the cancer has spread to the bone marrow, a bone marrow aspiration is needed to determine the progression of the disease.
Other inspections
● Perform the following tests before treatment with anthracyclines:
- electrocardiography
- Echocardiography or resting radionuclide ejection fraction scans
● Do the following tests before cisplatin treatment:
- Baseline hearing test
- creatinine clearance
Clinical Management
Treatment options for neuroblastoma depend on factors such as the size of the tumor, the extent of tumor spread, the site of origin of the tumor, the patient's age, pathological features, and the chromosomal and genetic variability of the tumor. Doctors use a comprehensive assessment of these factors to arrive at the patient's cancer risk classification (factors such as the chromosomal and genetic variability of the tumor. Through the comprehensive assessment of these factors, the doctor will arrive at the classification of the patient's cancer risk (very low risk, low risk, medium risk, high risk), and accordingly formulate a personalized treatment plan including surgery, chemotherapy, radiotherapy, autologous hematopoietic stem cell transplantation, immunotherapy, and its combination therapy.
Once a diagnosis has been made and tests completed, the patient and family are guided through the diagnosis and treatment.
Once a treatment plan is developed, chemotherapy is usually administered in an inpatient setting.
After completing the treatment cycle, patients were discharged home with detailed home care instructions and outpatient follow-up.
In high-risk patients, the feasibility of autologous HSCT should be evaluated early in the treatment planning phase.
Children with neuroblastoma should be admitted to the hospital to expedite diagnostic testing when their condition is unstable or when symptoms are evident.
Treatment strategies for the very-low-risk and low-risk groups
Patients in this group are usually treated with surgical removal of the tumor only. If the patient has significant symptoms, or if the tumor cannot be completely removed by surgery, it may be combined with adjuvant chemotherapy. For some patients with unresectable tumors, chemotherapy alone may be used.
Radiotherapy is usually not required in this group, but may be an option if the tumor is unresectable and chemotherapy is not effective, or if the patient develops symptoms of spinal cord compression or respiratory distress syndrome.
If the children in this group are under 1 year of age and asymptomatic, or under 6 months of age and have only smaller adrenal tumors, then the tumors may spontaneously regress, and therefore can usually be closely monitored without the urgency of surgery or chemotherapy.
Treatment strategies for the intermediate-risk group
The primary treatment for this group of patients is surgery and chemotherapy (4-8 courses). If the tumor is unresectable, it is also possible to have chemotherapy only, or chemotherapy followed by surgery after the tumor has shrunk.
For infants under 1 year of age, the necessity of chemotherapy is controversial, and surgery and postoperative observation may be the only option.
Radiotherapy may be an option if the tumor cannot be removed and chemotherapy is not effective, or if the patient has an emergency such as spinal cord compression or respiratory distress syndrome.
Treatment strategies for high-risk groups
The treatment of this group of patients was divided into three phases: induction, consolidation and maintenance.
Treatment in the induction phase was surgery and multi-drug chemotherapy. For patients who cannot have their tumors completely removed surgically, a second surgery is required after the chemotherapy has shrunk the tumor, although the extent and effectiveness of surgical removal is still controversial and usually requires a doctor's judgment based on actual circumstances and experience.
Meanwhile, during the induction phase, patients undergo a procedure to filter and collect hematopoietic stem cells from their blood in order to store their own hematopoietic stem cells for subsequent autologous hematopoietic stem cell transplantation during the consolidation phase.
Treatment during the consolidation phase is high-dose myeloablative chemotherapy, radiation therapy, and autologous peripheral blood HSCT. Myeloablative chemotherapy kills tumor cells and hematopoietic stem cells in the patient's body through high-dose chemotherapy. Radiotherapy, which is usually given after myeloablative chemotherapy, targets the primary site and persistent metastases, further killing residual tumor cells. Subsequently, during autologous hematopoietic stem cell transplantation, hematopoietic stem cells stored during the induction phase are injected into the patient's body to form new, healthy blood cells.
The current international recommendation for standard therapy in the high-risk group is two consecutive rounds of myeloablative chemotherapy and autologous hematopoietic stem cell transplantation. Two consecutive rounds of myeloablative chemotherapy and transplantation can achieve better treatment outcomes and significantly improve patient survival compared to a single round of myeloablative chemotherapy and transplantation. Two rounds of myeloablative chemotherapy and autologous HSCT are usually performed sequentially after 8 courses of induction chemotherapy in high-risk patients. If the patient is not ready for stem cell transplantation after 8 courses, chemotherapy can be continued to 12 courses.
The main purpose of the maintenance phase is to eliminate the residual tumor cells in the patient's body, which is treated by oral administration of isotretinoin. According to current research, the use of isotretinoin in the maintenance phase can help reduce the recurrence rate.
cutting-edge treatment
GD2 Antibody
The use of GD2 antibodies directed against neuroblastoma antigens is a new approach to treating high-risk neuroblastoma.GD2 is a molecule that is highly expressed in tumor cells in neuroblastoma. Because this molecule is found in much greater amounts on neuroblastoma than in normal cells, it can be used as a target to treat neuroblastoma.
The GD2 antibody is usually used along with isotretinoin in the maintenance phase of high-risk neuroblastoma, which can significantly improve patient survival, although there are some side effects - it can cause fever, nerve pain, and infusion reactions. In addition, the GD2 antibody has some neurotoxicity and may cause eye damage (e.g., dilated pupils, dulled light reflexes, and possibly photophobia) in a small number of patients.
There are currently two GD2 antibodies approved for marketing abroad, one in the United States and the other in Europe, both for the treatment of high-risk neuroblastoma. The U.S. version is called Dinutuximab (trade name Unituxin) and the European version is called Dinutuximab beta (trade name Qarziba).
Theoretically, these two versions of the GD2 antibody are actually one and the same thing, but due to differences in manufacturers and processes, the dosage and use of the two versions are different, and it is important to follow your doctor's instructions when using them. At present, both GD2 antibody drugs are not yet available in China, and both are on the State Food and Drug Administration's list of overseas marketed new drugs in urgent clinical need.
Iodine 131-MIBG therapy
When 131 Iodine-MIBG enters the bloodstream, it goes into neuroblastoma cells everywhere in the body. Because 131 iodine-MIBG is radioactive, it can precisely kill tumor cells. The use of this therapy in combination with other therapies is currently under investigation.
ALK inhibitor
In some patients with neuroblastoma, there is an abnormality in the ALK gene, and treatment with an ALK inhibitor (e.g., crizotinib, loratinib, etc.) may be attempted.
Side effects of treatment
Cancer treatments can cause side effects because treating tumor cells can also affect healthy tissue or organs, and neuroblastoma is no exception.
Side effects of chemotherapy depend on the type and dosage of the drug given and the length of time it is taken. Common side effects can include: hair loss, mouth sores, loss of appetite, nausea and vomiting, diarrhea or constipation, increased chance of infection, easy bruising or bleeding, and fatigue. The severity of side effects is related to the type of drug, the dose and the duration of chemotherapy. Usually, these side effects go away after the treatment is over.
Side effects of radiation therapy include fatigue, mild skin discomfort, nausea, and diarrhea. At the same time, children's bodies are often very sensitive to radiation therapy, which may increase the risk of distant secondary tumors. Radiation therapy to the brain and reproductive system may sometimes affect children's growth and development, so the radiation dose needs to be predicted in relation to the desired treatment goals.
In high-risk neuroblastoma survivors, growth retardation, thyroid dysfunction, learning disabilities, and secondary tumors may occur.
Information about the side effects of cancer treatment can be found in the Symptom Management in Children's Palliative and Hospice Care section; in addition to the early side effects of treatment, distant side effects may still occur after treatment is completed, as described in the Late Effects of Pediatric Oncology Treatment section.
Review and Follow-up
1. Monitoring of complications
After each course of treatment, complications should be monitored regularly and response to treatment assessed with diagnostic imaging, with myelosuppression and pancytopenia being common complications. If necessary, routine blood tests (with emphasis on platelet counts) should be performed twice weekly.
2. Detection of drug side effects
Some drugs (e.g., cisplatin, carboplatin, isocyclophosphamide) may affect liver and kidney function and require regular monitoring. Gastrointestinal symptoms caused by chemotherapy may affect diet. Therefore, close monitoring of electrolytes and oral electrolyte supplementation if necessary is required. Seek medical attention as soon as possible when hemoglobin drops to less than 8 g/d L, platelet count drops to less than 10,000, or any signs of bleeding are present.
Prognosis
In the United States, the overall 5-year survival rate for patients with neuroblastoma is 80%. The 5-year survival rate is 90% for young children under 1 year of age, 68% for children 1-4 years of age, 52% for children 5-9 years of age, and 46% for adolescents 10-21 years of age.
When looking at risk subgroups, the overall 5-year survival rate in the United States is above 95% in the very-low-risk and low-risk groups, 90-95% in the intermediate-risk group, and 40-50% in the high-risk group.
Improvements in diagnostic imaging modalities, medical and surgical management, and supportive care contribute to survival.
Factors that influence prognosis include:
● Age at diagnosis, tumor stage and risk grouping, primary site of the tumor, degree of differentiation of the tumor cells, heterogeneity of the tumor, chromosomal and genetic variants in the tumor cells, spread of the tumor, rate of growth of the tumor, and response to treatment all affect the prognosis.
● Overall, patients who were younger at diagnosis had a relatively better prognosis.
● Highly differentiated tumors usually have a better prognosis than poorly differentiated tumors.
● Tumors originating in the mediastinum generally have a better prognosis than tumors from other sites. Tumors originating in the adrenal gland have a relatively less favorable prognosis.
● In high-risk patients whose tumors have metastasized to the bone marrow and have responded poorly to induction phase therapy, the prognosis is usually less favorable.
● The overall 5-year survival rate for recurrent neuroblastoma after treatment is 20%. Of these, the prognosis for recurrence is better in very-low-risk, low-risk, and intermediate-risk patients, and less favorable in high-risk recurrences.
● The prognosis for recurrence is usually relatively good if the time of recurrence is more than one year from the last conjunctive treatment.
Follow-up & Review
follow up
After treatment is completed, follow-up should be performed and any signs or symptoms of recurrent disease should be closely monitored. This includes regular monitoring of urinary catecholamines, physical examination, and imaging. Because most relapses occur within the first 2 years of treatment, most protocols recommend close follow-up during this time.
Patients with childhood neuroblastoma who are generally free of recurrence within 3 years are considered cured. However, cancer treatment affects normal tissues and organs and may have long-term side effects. Therefore, even if a child is cured, long-term follow-up is still needed to assess the effects of treatment on growth, development, and organ toxicity.
Routine
not have
Cutting-edge Therapeutic & Clinical Research
not have
References
References
1. https://www.cancer.gov/types/neuroblastoma/hp/neuroblastoma-treatment-pdq
2. https://www.cancer.gov/types/neuroblastoma/patient/neuroblastoma-treatment-pdq
References
1. https://www.cancer.gov/types/neuroblastoma/hp/neuroblastoma-treatment-pdq
2. https://www.cancer.gov/types/neuroblastoma/patient/neuroblastoma-treatment-pdq
3. https://www.cancer.net/cancer-types/neuroblastoma-childhood/view-all
References
1. https://www.cancer.gov/types/neuroblastoma/hp/neuroblastoma-treatment-pdq
2. https://www.cancer.gov/types/neuroblastoma/patient/neuroblastoma-treatment-pdq
3. https://www.cancer.net/cancer-types/neuroblastoma-childhood/view-all
References:
1. https://www.cancer.gov/types/neuroblastoma/hp/neuroblastoma-treatment-pdq
2. https://www.cancer.gov/types/neuroblastoma/patient/neuroblastoma-treatment-pdq
3. https://www.cancer.net/cancer-types/neuroblastoma-childhood/view-all
References
1. https://www.cancer.gov/types/neuroblastoma/hp/neuroblastoma-treatment-pdq
2. https://www.cancer.gov/types/neuroblastoma/patient/neuroblastoma-treatment-pdq
3. https://www.cancer.net/cancer-types/neuroblastoma-childhood/view-all
References
1. https://www.cancer.gov/types/neuroblastoma/hp/neuroblastoma-treatment-pdq
2. https://www.cancer.gov/types/neuroblastoma/patient/neuroblastoma-treatment-pdq
3. https://www.cancer.net/cancer-types/neuroblastoma-childhood/view-all
4. https://www.cancer.gov/types/childhood-cancers/late-effects-hp-pdq
Audit specialists
Wang Huanmin, Director of Surgical Oncology, Beijing Children's Hospital, Capital Medical University
Personal Profile
https://baike.baidu.com/item/%E7%8E%8B%E7%84%95%E6%B0%91/17680619#viewPageContent
List of volunteers
Search
Related Articles

Relaxation Therapy & Peace Care
Jul 03, 2025

Rare Childhood Tumour
Jul 03, 2025

Inflammatory Myofibroblastoma
Jul 03, 2025

Langerhans Cell Histiocytosis
Jul 03, 2025

Angeioma
Jul 03, 2025